
Tuesday, December 27, 2011
Genome tree of life is largest yet for seed plants
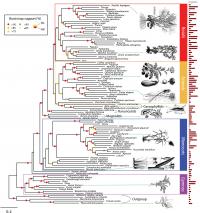
Scientists at the American Museum of Natural History, Cold Spring Harbor Laboratory, The New York Botanical Garden, and New York University have created the largest genome-based tree of life for seed plants to date. Their findings, published today in the journal PLoS Genetics, plot the evolutionary relationships of 150 different species of plants based on advanced genome-wide analysis of gene structure and function. This new approach, called "functional phylogenomics," allows scientists to reconstruct the pattern of events that led to the vast number of plant species and could help identify genes used to improve seed quality for agriculture.
more
Friday, December 9, 2011
KABOOM! A new suffix array based algorithm for clustering expression data
Abstract
Motivation: Second-generation sequencing technology has reinvigorated research using expression data, and clustering such data remains a significant challenge, with much larger datasets and with different error profiles. Algorithms that rely on all-versus-all comparison of sequences are not practical for large datasets.
Results: We introduce a new filter for string similarity which has the potential to eliminate the need for all-versus-all comparison in clustering of expression data and other similar tasks. Our filter is based on multiple long exact matches between the two strings, with the additional constraint that these matches must be sufficiently far apart. We give details of its efficient implementation using modified suffix arrays. We demonstrate its efficiency by presenting our new expression clustering tool, wcd-express, which uses this heuristic. We compare it to other current tools and show that it is very competitive both with respect to quality and run time.
Availability: Source code and binaries available under GPL athttp://code.google.com/p/wcdest. Runs on Linux and MacOS X.
Saturday, December 3, 2011
The Infobiotics Workbench: an integrated in silico modelling platform for Systems and Synthetic Biology
Abstract
Summary: The Infobiotics Workbench is an integrated software suite incorporating model specification, simulation, parameter optimization and model checking for Systems and Synthetic Biology. A modular model specification allows for straightforward creation of large-scale models containing many compartments and reactions. Models are simulated either using stochastic simulation or numerical integration, and visualized in time and space. Model parameters and structure can be optimized with evolutionary algorithms, and model properties calculated using probabilistic model checking.
Availability: Source code and binaries for Linux, Mac and Windows are available at http://www.infobiotics.org/infobiotics-workbench/; released under the GNU General Public License (GPL) version 3.
Saturday, November 26, 2011
Nano-ultra high performance liquid chromatography

Implementation of the Thermo Scientific EASY-nLC 1000 has enabled 34 percent more peptide and 22 percent more protein identifications within the CEBI laboratory, significantly increasing productivity. Additionally, the instrument has improved separations and facilitated the analysis of more complex samples within the laboratory.
Based in Odense, the University of Southern Denmark’s Department of Biochemistry and Molecular Biology is world-renowned in the field of proteomics, with research focusing on gene structure and organization, regulation and RNA translation. CEBI is a specialist proteomics research laboratory, aimed at applying modern methods of mass spectrometry and proteomics to functional analysis of genes.
Prior to implementing nano-UHPLC technology, CEBI used conventional HPLC. However they needed a more complete solution, such as EASY-nLC 1000, to accelerate productivity and improve efficiencies. The EASY-nLC 1000 is used in conjunction with Thermo Scientific ion trap and Orbitrap mass spectrometers for a wide range of proteomics research, including the study of complex mouse dendritic cells.
Using the EASY-nLC 1000 has allowed CEBI to take advantage of the ability to facilitate dedicated separation of biomolecules at ultra-high pressures. The higher operating pressures provided by the EASY-nLC 1000 have increased the number of peptide and protein identifications performed within the laboratory by 34 percent and 22 percent respectively. The instrument also allows selection of the most appropriate column dimensions and solid phase materials in order to identify the most suitable configuration to match analytical requirements. Using thinner, longer columns and smaller beads has improved separation and ionization, enhancing sensitivity and allowing the CEBI team to scale down experiments and reduce costs.
“The EASY-nLC 1000 is a highly robust instrument offering nano-UHPLC performance dedicated to proteomics scientists,” said Lasse Falkenby, research assistant at CEBI. “We decided to implement the new system in our laboratory as we felt that it would revolutionise the way we perform LC-MS analyses. The instrument has allowed our laboratory to increase productivity considerably in a way that would never have been possible before. Implementation was a very smooth process requiring minimal configuration effort and the Thermo Fisher Scientific team of experts provided valuable support both throughout and after this process.”
For more information, visit www.thermoscientific.com/easylc
Thursday, November 17, 2011
Pico Computing Demonstrates Bioinformatics Acceleration at SC 2011
Pico's SC5 FPGA cluster reduces short read sequencing from 6 1/2 hours to just one minute
Seattle, WA (PRWEB) November 09, 2011
Pico Computing will be demonstrating an FPGA implementation of BFAST resulting in a 350X acceleration over a software-only implementation running on two quad core Intel Xeon processors. The 350X acceleration was achieved using 8 Pico M-503 FPGA modules in their SC5 SuperCluster chassis. The BFAST algorithm is primarily used in short read genome mapping.
Read more: http://www.sfgate.com/cgi-bin/article.cgi?f=/g/a/2011/11/09/prweb8945226.DTL#ixzz1dyI5LQs
Monday, November 7, 2011
Gene Ontology-driven inference of protein-protein interactions using inducers

Motivation: Protein-protein interactions (PPI) are pivotal for many biological processes and similarity in Gene Ontology (GO) annotation has been found to be one of the strongest indicators for PPI. Most GO-driven algorithms for PPI inference combine machine learning and semantic similarity techniques. We introduce the concept of inducers as a method to integrate both approaches more effectively, leading to superior prediction accuracies.
Results: An inducer (ULCA) in combination with a Random Forest classifier compares favorably to several sequenced-based methods, semantic similarity measures and multi-kernel approaches. On a newly created set of high-quality interaction data, the proposed method achieves high cross-species prediction accuracies (AUC ≤ 0.88), rendering it a valuable companion to sequence-based methods.
Availability: Software and datasets are available athttp://bioinformatics.org.au/go2ppi/
Saturday, October 29, 2011
Enhanced peptide quantification using spectral count clustering and cluster abundance
Quantification of protein expression by means of mass spectrometry (MS) has been introduced in various proteomics studies. In particular, two label-free quantification methods, such as spectral counting and spectra feature analysis have been extensively investigated in a wide variety of proteomic studies.
The cornerstone of both methods is peptide identification based on a proteomic database search and subsequent estimation of peptide retention time. However, they often suffer from restrictive database search and inaccurate estimation of the liquid chromatography (LC) retention time.
Furthermore, conventional peptide identification methods based on the spectral library search algorithms such as SEQUEST or SpectraST have been found to provide neither the best match nor high-scored matches. Lastly, these methods are limited in the sense that target peptides cannot be identified unless they have been previously generated and stored into the database or spectral libraries.To overcome these limitations, we propose a novel method, namely Quantification method based on Finding the Identical Spectral set for a Homogenous peptide (Q-FISH) to estimate the peptide's abundance from its tandem mass spectrometry (MS/MS) spectra through the direct comparison of experimental spectra.
Intuitively, our Q-FISH method compares all possible pairs of experimental spectra in order to identify both known and novel proteins, significantly enhancing identification accuracy by grouping replicated spectra from the same peptide targets.
Results: We applied Q-FISH to Nano-LC-MS/MS data obtained from human hepatocellular carcinoma (HCC) and normal liver tissue samples to identify differentially expressed peptides between the normal and disease samples. For a total of 44,318 spectra obtained through MS/MS analysis, Q-FISH yielded 14,747 clusters.
Among these, 5,777 clusters were identified only in the HCC sample, 6,648 clusters only in the normal tissue sample, and 2,323 clusters both in the HCC and normal tissue samples. While it will be interesting to investigate peptide clusters only found from one sample, further examined spectral clusters identified both in the HCC and normal samples since our goal is to identify and assess differentially expressed peptides quantitatively.
The next step was to perform a beta-binomial test to isolate differentially expressed peptides between the HCC and normal tissue samples. This test resulted in 84 peptides with significantly differential spectral counts between the HCC and normal tissue samples.
We independently identified 50 and 95 peptides by SEQUEST, of which 24 and 56 peptides, respectively, were found to be known biomarkers for the human liver cancer. Comparing Q-FISH and SEQUEST results, we found 22 of the differentially expressed 84 peptides by Q-FISH were also identified by SEQUEST.
Remarkably, of these 22 peptides discovered both by Q-FISH and SEQUEST, 13 peptides are known for human liver cancer and the remaining 9 peptides are known to be associated with other cancers.
Conclusions: We proposed a novel statistical method, Q-FISH, for accurately identifying protein species and simultaneously quantifying the expression levels of identified peptides from mass spectrometry data. Q-FISH analysis on human HCC and liver tissue samples identified many protein biomarkers that are highly relevant to HCC.
Q-FISH can be a useful tool both for peptide identification and quantification on mass spectrometry data analysis. It may also prove to be more effective in discovering novel protein biomarkers than SEQUEST and other standard methods.
Author: Seungmook LeeMin-Seok KwonHyoung-Joo LeeYoung-Ki PaikHaixu TangJae LeeTaesung Park
Credits/Source: BMC Bioinformatics 2011, 12:423
Friday, October 21, 2011
Researchers generate first complete 3-D structures of bacterial chromosome
A team of researchers at the University of Massachusetts Medical School, Harvard Medical School, Stanford University and the Prince Felipe Research Centre in Spain have deciphered the complete three-dimensional structure of the bacterium Caulobacter cresc ...
Monday, October 17, 2011
A QuantuMDx Leap for Handheld DNA Sequencing
"October 17, 2011 | MONTREAL – Speaking for the first time in his life as a commercial consultant rather than a public servant, Sir John Burn, a highly respected clinical geneticist in the United Kingdom, provided the first glimpse at a nanowire technology for rapid DNA genotyping that could eventually mature into the world’s first handheld DNA sequencer.
Burn previewed a potentially disruptive genome diagnostic technology in a presentation on the closing day of the International Congress of Human Genetics in Montreal last weekend.
One day a week, Burn, professor of clinical genetics at Newcastle University, serves as medical director for QuantuMDx (QMDx), a British start-up co-founded by molecular biologist Jonathan O’Halloran and healthcare executive Elaine Warburton."
Tuesday, October 11, 2011
Drug2Gene
Saturday, October 8, 2011
Gamers succeed where scientists fail
"Gamers have solved the structure of a retrovirus enzyme whose configuration had stumped scientists for more than a decade. The gamers achieved their discovery by playing Foldit, an online game that allows players to collaborate and compete in predicting the structure of protein molecules."

more
more
Wednesday, October 5, 2011
mz5: Space- and time-efficient storage of mass spectrometry data sets
"Across a host of mass spectrometry (MS)-driven -omics fields, researchers witness the acquisition of ever increasing amounts of high throughput MS datasets and the need for their compact yet efficiently accessible storage has become clear.
The HUPO proteomics standard initiative (PSI) has defined an ontology and associated controlled vocabulary that specifies the contents of MS data files in terms of an open data format. Current implementations are the mzXML and mzML formats (mzML specification), both of which are based on an XML representation of the data. As a consequence, these formats are not particular efficient with respect to their storage space requirements or I/O performance.
This contribution introduces mz5, an implementation of the PSI mzML ontology that is based on HDF5, an efficient, industrial strength storage backend.
Compared to the current mzXML and mzML standards, this strategy yields an average file size reduction of a factor of ~2 and increases I/O performace ~3-4 fold.
The format is implemented as part of the ProteoWizard project."
more
The HUPO proteomics standard initiative (PSI) has defined an ontology and associated controlled vocabulary that specifies the contents of MS data files in terms of an open data format. Current implementations are the mzXML and mzML formats (mzML specification), both of which are based on an XML representation of the data. As a consequence, these formats are not particular efficient with respect to their storage space requirements or I/O performance.
This contribution introduces mz5, an implementation of the PSI mzML ontology that is based on HDF5, an efficient, industrial strength storage backend.
Compared to the current mzXML and mzML standards, this strategy yields an average file size reduction of a factor of ~2 and increases I/O performace ~3-4 fold.
The format is implemented as part of the ProteoWizard project."
more
Friday, September 23, 2011
Gamers Unlock Protein Mystery That Baffled AIDS Researchers For Years

"In just three weeks, gamers deciphered the structure of a key protein in the development of AIDS that has stumped scientists for years. According to a studypublished Sunday in the journal Nature Structural & Molecular Biology, the findings could present a significant breakthrough for AIDS and HIV research.
Using an online game called Foldit, players were able to predict the structure of a protein called retroviral protease, an enzyme that plays a critical role in the way HIV multiplies. Unlocking the build of the protein could theoretically aid scientists in developing drugs that would stop protease from spreading.
Following the failure of a wide range of attempts to solve the crystal structure of M-PMV retroviral protease by molecular replacement, we challenged players of the protein folding game Foldit to produce accurate models of the protein,” the study reads. “Remarkably, Foldit players were able to generate models of sufficient quality for successful molecular replacement and subsequent structure determination. The refined structure provides new insights for the design of antiretroviral drugs.”
Developed by researchers at the University of Washington, Foldit turns scientific problems into competitive games. Players were charged with using spatial and critical thinking skills to build 3D models of protease. Few of these players had any kind of background in biochemistry.
According to Fox, it took players a matter of days to come up with models that were solid enough for researchers to translate into scientific rendering of the protein.
“People have spatial reasoning skills, something computers are not yet good at,” Foldit’s lead designer Seth Cooper said in a statement. “Games provide a framework for bringing together the strengths of computers and humans.”
Friday, September 2, 2011
HPLC 2011 highlights chromatography technologies
Thermo Fisher Scientific is to highlight its expanded offering of chromatography instruments, software and consumables, including the Accucore HPLC column range, at the HPLC 2011 event in Budapest.
The Accucore HPLC column range is said to enhance laboratory workflow and efficiency by providing increased sensitivity and peak resolution in columns that are compatible with almost any instrument.
Thermo Fisher Scientific will also showcase the Easy-NLC 1000 split-free nano-flow system for advanced proteomics research at HPLC 2011.
This system is said to increase chromatographic resolution and, as a result, protein and peptide identifications, with ultra-high-pressure operation.
The company will also highlight the Velos Pro linear ion trap and the Orbitrap Velos Pro hybrid FTMS mass spectrometers.
These systems are said to provide improved quantitative performance, faster scanning, trap higher energy collision dissociation (HCD) and enhanced robustness.
Thermo Fisher Scientific will also introduce the Q Exactive high-performance benchtop quadrupole-Orbitrap LC-MS/MS, which combines quadrupole precursor selection and high-resolution accurate mass (HR/AM) Orbitrap mass analysis to deliver high-confidence quantitative and qualitative workflows.
With the HR/AM Quanfirmation capability, the Q Exactive mass spectrometer can identify, quantify and confirm more trace-level peptides and proteins in complex mixtures in one analytical run.
The Orbitrap Elite hybrid mass spectrometer is said to provide the resolution and sensitivity required to improve the determination of the molecular weights of intact proteins within laboratories, as well as enable greater proteome coverage through improved protein, PTM and peptide identification, even at low abundances.
Read more: http://www.laboratorytalk.com/news/tel/tel185.html#ixzz1RfGDUTMa
CloVR

"Background
Next-generation sequencing technologies have decentralized sequence acquisition, increasing the demand for new bioinformatics tools that are easy to use, portable across multiple platforms, and scalable for high-throughput applications. Cloud computing platforms provide on-demand access to computing infrastructure over the Internet and can be used in combination with custom built virtual machines to distribute pre-packaged with pre-configured software.
Results
We describe the Cloud Virtual Resource, CloVR, a new desktop application for push-button automated sequence analysis that can utilize cloud computing resources. CloVR is implemented as a single portable virtual machine (VM) that provides several automated analysis pipelines for microbial genomics, including 16S, whole genome and metagenome sequence analysis. The CloVR VM runs on a personal computer, utilizes local computer resources and requires minimal installation, addressing key challenges in deploying bioinformatics workflows. In addition CloVR supports use of remote cloud computing resources to improve performance for large-scale sequence processing. In a case study, we demonstrate the use of CloVR to automatically process next-generation sequencing data on multiple cloud computing platforms.
Conclusion
The CloVR VM and associated architecture lowers the barrier of entry for utilizing complex analysis protocols on both local single- and multi-core computers and cloud systems for high throughput data processing."
Monday, August 29, 2011
New Roles Emerge For Non-Coding RNAs In Directing Embryonic Development
Scientists at the Broad Institute of MIT and Harvard have discovered that a mysterious class of large RNAs plays a central role in embryonic development, contrary to the dogma that proteins alone are the master regulators of this process. The research, published online August 28 in the journal Nature, reveals that these RNAs orchestrate the fate of embryonic stem (ES) cells by keeping them in their fledgling state or directing them along the path to cell specialization.
Broad scientists discovered several years ago that the human and mouse genomes encode thousands of unusual RNAs — termed large, intergenic non-coding RNAs (lincRNAs) —but their role was almost entirely unknown. By studying more than 100 lincRNAs in ES cells, the researchers now show that these RNAs help regulate development by physically interacting with proteins to coordinate gene expression and suggest that lincRNAs may play similar roles in most cells.
Tuesday, August 23, 2011
Monday, August 22, 2011
Comparative analysis of algorithms for next-generation sequencing read alignment
Abstract
Motivation: The advent of next-generation sequencing (NGS) techniques presents many novel opportunities for many applications in life sciences. The vast number of short reads produced by these techniques, however, pose significant computational challenges. The first step in many types of genomic analysis is the mapping of short reads to a reference genome, and several groups have developed dedicated algorithms and software packages to perform this function. As the developers of these packages optimize their algorithms with respect to various considerations, the relative merits of different software packages remain unclear. However, for scientists who generate and use NGS data for their specific research projects, an important consideration is choosing the software that is most suitable for their application.
Results: With a view to comparing existing short read alignment software, we develop a simulation and evaluation suite, SEAL, which simulates NGS runs for different configurations of various factors, including sequencing error, indels, and coverage. We also develop criteria to compare the performances of software with disparate output structure (e.g., some packages return a single alignment while some return multiple possible alignments). Using these criteria, we comprehensively evaluate the performances of Bowtie, BWA, mr- and mrsFAST, Novoalign, SHRiMP and SOAPv2, with regard to accuracy and runtime.
Monday, August 15, 2011
Can the false-discovery rate be misleading?
"The decoy-database approach is currently the gold standard for assessing the confidence of identifications in shotgun proteomic experiments. Here we demonstrate that what might appear to be a good result under the decoy-database approach for a given false-discovery rate could be, in fact, the product of overfitting. This problem has been overlooked until now and could lead to obtaining boosted identification numbers whose reliability does not correspond to the expected false-discovery rate. To remedy this, we are introducing a modified version of the method, termed a semi-labeled decoy approach, which enables the statistical determination of an overfitted result."
more
Comments: Dr. Pavel Pevzner published some paper with similar idea, if my memory services me correct.
Monday, August 8, 2011
Bioinformatics for Cancer Genomics (BiCG)
Workshop Details
Date: August 29 - September 2, 2011
Location: MaRS Building, Downtown Toronto
Lead Faculty (2011): John McPherson, Francis Ouellette, Paul Boutros, Michael Brudno, Sohrab Shah, Gary Bader and Anna Lapuk
Registration Fee for Applications received before July 29, 2011: $950 + HST
Registration Fee for Applications received after July 29, 2011: $1150 + HST
Location: MaRS Building, Downtown Toronto
Lead Faculty (2011): John McPherson, Francis Ouellette, Paul Boutros, Michael Brudno, Sohrab Shah, Gary Bader and Anna Lapuk
Registration Fee for Applications received before July 29, 2011: $950 + HST
Registration Fee for Applications received after July 29, 2011: $1150 + HST
Awards available for 2011.
Course Objectives
Cancer research has rapidly embraced high throughput technologies into its research, using various microarray, tissue array, and next generation sequencing platforms. The result has been a rapid increase in cancer data output and data types. Now more than ever, having the informatic skills and knowledge of available bioinformatic resources specific to cancer is critical.
Cancer research has rapidly embraced high throughput technologies into its research, using various microarray, tissue array, and next generation sequencing platforms. The result has been a rapid increase in cancer data output and data types. Now more than ever, having the informatic skills and knowledge of available bioinformatic resources specific to cancer is critical.
The CBW will host a 5-day workshop covering the key bioinformatics concepts and tools required to analyze cancer genomic data sets. Participants will gain experience in genomic data visualization tools which will be applied throughout the development of the skills required to analyze cancer -omic data for gene expression, genome rearrangement, somatic mutations and copy number variation. The workshop will conclude with analyzing and conducting pathway analysis on the resultant cancer gene list and integration of clinical data.
Monday, August 1, 2011
Ontario Institute for Cancer Research Receives $420 Million Over Five Years for Important Cancer Research in Ontario Read more: Ontario Institute for Cancer Research Receives $420 Million Over Five Years for Important Cancer Research in Ontario
TORONTO, July 26, 2011 /PRNewswire/ -- The Ontario Institute for Cancer Research (OICR) will receive $420 million over five years from the Government of Ontario to continue its research into the prevention, early detection, diagnosis and treatment of cancer. The Institute will also occupy two floors of Phase II of the MaRS Centre in addition to its current laboratories and offices at its headquarters in the MaRS Centre. The announcement was made today by Dr. Tom Hudson, President and Scientific Director.
Today's announcement means continued support for the Institute in fulfilling the ambitious goals set out in its second Strategic Plan for 2010-2015, which focuses on the adoption of more personalized approaches to cancer diagnosis and treatment. OICR's current research activities in genomics and bioinformatics will be expanded in the new space in Phase II, allowing the Institute to increase its capacity to make new discoveries and move them out of the laboratory into the clinic for the benefit of patients. The funding will also enable the Ontario Health Study to complete its recruitment plans for the Study which will lead to better prevention of cancer and other chronic diseases.
Read more: Ontario Institute for Cancer Research Receives $420 Million Over Five Years for Important Cancer Research in Ontario - FierceBiotech http://www.fiercebiotech.com/press-releases/ontario-institute-cancer-research-receives-420-million-over-five-years-impo#ixzz1TnYjBOiS
Saturday, July 23, 2011
A lack of structure facilitates protein synthesis
Texts without spaces are not very legible, as they make it very difficult for the reader to identify where a word begins and where it ends. When genetic information in our cells is read and translated into proteins, the enzymes responsible for this task face a similar challenge. They must find the correct starting point for protein synthesis. Therefore, in organisms with no real nucleus, a point exists shortly before the start codon, to which the enzymes can bind particularly well. This helps them find the starting point itself. However, genes that do not have this sequence are also reliably translated into proteins. Scientists from the Max Planck Institute of Molecular Plant Physiology in Potsdam have discovered that the structure of the messenger RNA probably plays a crucial role in this process.

Saturday, July 16, 2011
Predicting liver transplant rejection
"The survival rate of liver transplant patients one year after treatment has improved from about 30% in the 1970s to more than 80%, with acute cellular rejection (ACR) the most common complication. It occurs in about 30% of cases and is generally arrested by drug treatment. However, if ACR occurs more than one year after the transplant, survival rates plummet.
The diagnosis of ACR requires a tissue biopsy, which is risky and uncomfortable for the patient. Even then, the interpretation of samples is difficult because the three clinical predictors are not always present.
These problems have prompted scientists in the US to look for a non-invasive alternative diagnosis for ACR and they turned to proteomics for the solution. Michael Charlton and colleagues from the Mayo Clinic and Foundation, Rochester, MN, and the University of Alabama at Birmingham decided to look at the serum proteome to see if any indicators of ACR were detectable.
Human serum contains many high-abundant proteins but, if they are removed before protein analysis, it is possible to detect low-abundant proteins which are transiently present in serum.
So, proteins secreted by cells or produced during cell destruction become visible. These include hormones and cytokines which are transported in serum to their destinations within the human body."

Subscribe to:
Posts (Atom)