
Showing posts with label genomics. Show all posts
Showing posts with label genomics. Show all posts
Tuesday, December 27, 2011
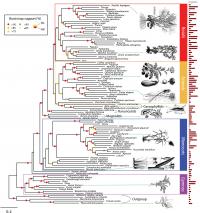
Genome tree of life is largest yet for seed plants
Scientists at the American Museum of Natural History, Cold Spring Harbor Laboratory, The New York Botanical Garden, and New York University have created the largest genome-based tree of life for seed plants to date. Their findings, published today in the journal PLoS Genetics, plot the evolutionary relationships of 150 different species of plants based on advanced genome-wide analysis of gene structure and function. This new approach, called "functional phylogenomics," allows scientists to reconstruct the pattern of events that led to the vast number of plant species and could help identify genes used to improve seed quality for agriculture.
more
Monday, November 7, 2011
Gene Ontology-driven inference of protein-protein interactions using inducers

Motivation: Protein-protein interactions (PPI) are pivotal for many biological processes and similarity in Gene Ontology (GO) annotation has been found to be one of the strongest indicators for PPI. Most GO-driven algorithms for PPI inference combine machine learning and semantic similarity techniques. We introduce the concept of inducers as a method to integrate both approaches more effectively, leading to superior prediction accuracies.
Results: An inducer (ULCA) in combination with a Random Forest classifier compares favorably to several sequenced-based methods, semantic similarity measures and multi-kernel approaches. On a newly created set of high-quality interaction data, the proposed method achieves high cross-species prediction accuracies (AUC ≤ 0.88), rendering it a valuable companion to sequence-based methods.
Availability: Software and datasets are available athttp://bioinformatics.org.au/go2ppi/
Monday, October 17, 2011
A QuantuMDx Leap for Handheld DNA Sequencing
"October 17, 2011 | MONTREAL – Speaking for the first time in his life as a commercial consultant rather than a public servant, Sir John Burn, a highly respected clinical geneticist in the United Kingdom, provided the first glimpse at a nanowire technology for rapid DNA genotyping that could eventually mature into the world’s first handheld DNA sequencer.
Burn previewed a potentially disruptive genome diagnostic technology in a presentation on the closing day of the International Congress of Human Genetics in Montreal last weekend.
One day a week, Burn, professor of clinical genetics at Newcastle University, serves as medical director for QuantuMDx (QMDx), a British start-up co-founded by molecular biologist Jonathan O’Halloran and healthcare executive Elaine Warburton."
Monday, August 29, 2011
New Roles Emerge For Non-Coding RNAs In Directing Embryonic Development
Scientists at the Broad Institute of MIT and Harvard have discovered that a mysterious class of large RNAs plays a central role in embryonic development, contrary to the dogma that proteins alone are the master regulators of this process. The research, published online August 28 in the journal Nature, reveals that these RNAs orchestrate the fate of embryonic stem (ES) cells by keeping them in their fledgling state or directing them along the path to cell specialization.
Broad scientists discovered several years ago that the human and mouse genomes encode thousands of unusual RNAs — termed large, intergenic non-coding RNAs (lincRNAs) —but their role was almost entirely unknown. By studying more than 100 lincRNAs in ES cells, the researchers now show that these RNAs help regulate development by physically interacting with proteins to coordinate gene expression and suggest that lincRNAs may play similar roles in most cells.
Monday, August 8, 2011
Bioinformatics for Cancer Genomics (BiCG)
Workshop Details
Date: August 29 - September 2, 2011
Location: MaRS Building, Downtown Toronto
Lead Faculty (2011): John McPherson, Francis Ouellette, Paul Boutros, Michael Brudno, Sohrab Shah, Gary Bader and Anna Lapuk
Registration Fee for Applications received before July 29, 2011: $950 + HST
Registration Fee for Applications received after July 29, 2011: $1150 + HST
Location: MaRS Building, Downtown Toronto
Lead Faculty (2011): John McPherson, Francis Ouellette, Paul Boutros, Michael Brudno, Sohrab Shah, Gary Bader and Anna Lapuk
Registration Fee for Applications received before July 29, 2011: $950 + HST
Registration Fee for Applications received after July 29, 2011: $1150 + HST
Awards available for 2011.
Course Objectives
Cancer research has rapidly embraced high throughput technologies into its research, using various microarray, tissue array, and next generation sequencing platforms. The result has been a rapid increase in cancer data output and data types. Now more than ever, having the informatic skills and knowledge of available bioinformatic resources specific to cancer is critical.
Cancer research has rapidly embraced high throughput technologies into its research, using various microarray, tissue array, and next generation sequencing platforms. The result has been a rapid increase in cancer data output and data types. Now more than ever, having the informatic skills and knowledge of available bioinformatic resources specific to cancer is critical.
The CBW will host a 5-day workshop covering the key bioinformatics concepts and tools required to analyze cancer genomic data sets. Participants will gain experience in genomic data visualization tools which will be applied throughout the development of the skills required to analyze cancer -omic data for gene expression, genome rearrangement, somatic mutations and copy number variation. The workshop will conclude with analyzing and conducting pathway analysis on the resultant cancer gene list and integration of clinical data.
Saturday, July 23, 2011
A lack of structure facilitates protein synthesis
Texts without spaces are not very legible, as they make it very difficult for the reader to identify where a word begins and where it ends. When genetic information in our cells is read and translated into proteins, the enzymes responsible for this task face a similar challenge. They must find the correct starting point for protein synthesis. Therefore, in organisms with no real nucleus, a point exists shortly before the start codon, to which the enzymes can bind particularly well. This helps them find the starting point itself. However, genes that do not have this sequence are also reliably translated into proteins. Scientists from the Max Planck Institute of Molecular Plant Physiology in Potsdam have discovered that the structure of the messenger RNA probably plays a crucial role in this process.

Sunday, June 5, 2011
PacBio RS Now Shipping!

It's official, Single Molecule Real Time (SMRT™) Sequencing is here! Last month we announced that we are now shipping commercial PacBio RS systems. This major milestone comes after more than 7 years of development. We at Pacific Biosciences have witnessed the power of our single molecule, real-time technology. Over the past year, it was even more thrilling to see top researchers from around the world present data taken from their new PacBio RS systems. Equally satisfying was observing the strong view among those customers that our technology will enable real applications that were not possible even six months ago. We are extremely excited to now deliver this technology out broadly to the world.
more
Tuesday, May 3, 2011
A window into third-generation sequencing

First- and second-generation sequencing technologies have led the way in revolutionizing the field of genomics and beyond, motivating an astonishing number of scientific advances, including enabling a more complete understanding of whole genome sequences and the information encoded therein, a more complete characterization of the methylome and transcriptome and a better understanding of interactions between proteins and DNA. Nevertheless, there are sequencing applications and aspects of genome biology that are presently beyond the reach of current sequencing technologies, leaving fertile ground for additional innovation in this space. In this review, we describe a new generation of single-molecule sequencing technologies (third-generation sequencing) that is emerging to fill this space, with the potential for dramatically longer read lengths, shorter time to result and lower overall cost.
more
Get SMRT: Pacific Biosciences Unveils Software Suite with Commercial Launch
April 29, 2011 | Third-generation sequencing company Pacific Biosciences (PacBio) began commercial shipment of its PacBio RS single-molecule sequencer this week. The instrument has been in beta testing at 11 institutions in North America and elsewhere for the past year. A notable success was the recent sequencing and identification of the cholera strain sweeping Haiti after the devastating 2010 earthquake.
In a briefing with Bio-IT World, PacBio staffers Kevin Corcoran, Jon Sorenson and Edwin Hauw previewed the new suite of software tools on the RS sequencer. The SMRT (single molecule/real time) Analysis software suite features web-based software, an analysis pipeline framework, and algorithms for sequence alignment and de novo assembly.
“We’re accelerating the development of software with the community,” says Kevin Corcoran. “A key feature of third-generation sequencing is that [the technology] doesn’t match up with what’s out there now. The key features of the PacBio system include fast time to result, high granularity, long read lengths, and new sequencing modes, including a circular mode and strobe sequencing.”
PacBio’s single-molecule sequencing system offers significantly longer read lengths (1,000 bases on average) than its second-generation sequencing rivals, and faster run times. That said, the total sequence throughput per run is currently less than other commercial platforms. The single-read accuracy hovers in the 85-90% range.
A revelatory feature of the SMRT software portal is that it captures kinetic information – the time for each registered nucleotide to be captured and incorporated into the growing DNA strand. “This is the first time you can watch DNA polymerase in real time, so that kinetic information will provide additional applications that have never been enabled before,” says Corcoran.
The genome browser is called SMRT View. “This takes advantage of our longer reads and kinetic information,” says Sorenson. It includes strobe and consensus sequence modes, allowing the user tovisualize and interact with secondary analysis sequence data. PacBio says the interactive graphical representations of variants, quality values, and other metrics is the first data visualization application that can visualize kinetics and structure information unique to PacBio's SMRT technology.

Wednesday, January 5, 2011
The complexity of gene expression dynamics revealed by permutation entropy

Background
High complexity is considered a hallmark of living systems. Here we investigate the complexity of temporal gene expression patterns using the concept of Permutation Entropy (PE) first introduced in dynamical systems theory. The analysis of gene expression data has so far focused primarily on the identification of differentially expressed genes, or on the elucidation of pathway and regulatory relationships. We aim to study gene expression time series data from the viewpoint of complexity.
Results
Applying the PE complexity metric to abiotic stress response time series data in Arabidopsis thaliana, genes involved in stress response and signaling were found to be associated with the highest complexity not only under stress, but surprisingly, also under reference, non-stress conditions. Genes with house-keeping functions exhibited lower PE complexity. Compared to reference conditions, the PE of temporal gene expression patterns generally increased upon stress exposure. High-complexity genes were found to have longer upstream intergenic regions and more cis-regulatory motifs in their promoter regions indicative of a more complex regulatory apparatus needed to orchestrate their expression, and to be associated with higher correlation network connectivity degree. Arabidopsis genes also present in other plant species were observed to exhibit decreased PE complexity compared to Arabidopsis specific genes.
Conclusions
We show that Permutation Entropy is a simple yet robust and powerful approach to identify temporal gene expression profiles of varying complexity that is equally applicable to other types of molecular profile data.
Thursday, November 25, 2010
Subscribe to:
Posts (Atom)